從結構到機制分類:用 AlphaMissense 和 FoldX 預測 TP53 突變是「顯性抑制型」還是「功能喪失型」——一個預先註冊的陰性結果
這篇文章在講什麼?
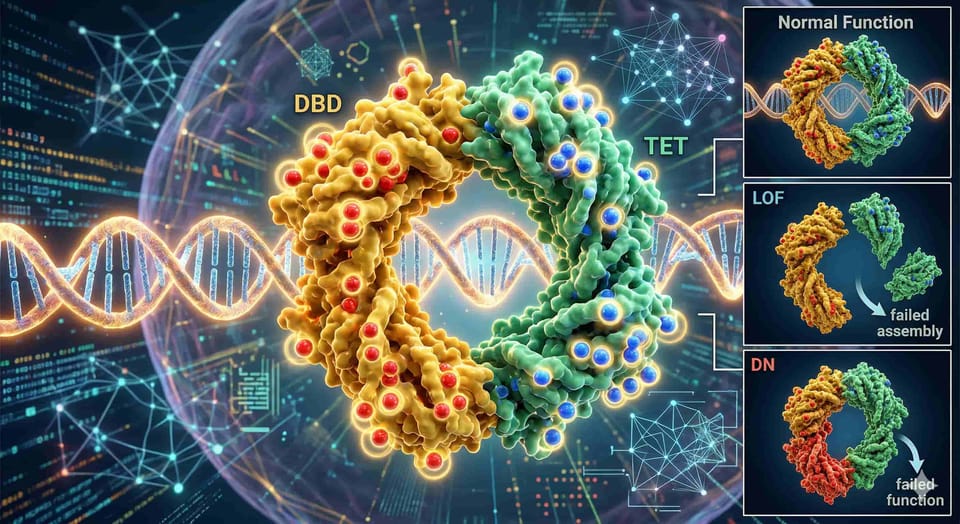
想像你的細胞裡有一個「基因守門人」叫做 TP53——它是人體最重要的腫瘤抑制蛋白,負責在 DNA 受損時踩煞車,阻止細胞癌變,TP53 工作時會四個一組(四聚體),像四個人手拉手圍成一圈才能發揮功能。
現在,如果其中一個 TP53 因為基因突變而壞掉了,會發生兩種截然不同的事:
功能喪失型(Loss-of-Function, LOF):壞掉的那一個自己退場,剩下三個正常的繼續工作,功能損失大約 25%。
顯性抑制型(Dominant-Negative, DN):壞掉的那一個不但不退場,還混進四聚體裡面搗亂,讓整組人都無法工作——一個老鼠屎壞了一鍋粥。
DN 突變的臨床後果比 LOF 嚴重得多,跟更高的癌症風險和更早的發病年齡有關,所以,能不能用機器學習來預測一個突變是 DN 還是 LOF?
這篇論文做了一件很少見的事:預先註冊假說、設定判定標準,然後誠實報告了一個陰性結果,作者假設「考慮蛋白質複合體結構的 FoldX 特徵」能超越簡單的 AlphaMissense + 單體 FoldX 基線模型,但結果是——沒有超越,AUROC 只提升了 0.013,信賴區間跨越零。
但這個「失敗」反而揭示了兩個意外發現:
- AlphaMissense + 單體 FoldX 這個簡單基線就達到了 AUROC 0.892,遠超預期
- 光靠「突變在蛋白質的哪個位置」這一個特徵,Random Forest 就達到 AUROC 0.946
這告訴我們:在 TP53 這個特定的資料集上,DN 和 LOF 突變的分佈本身就有極強的位置規律——這不是 bug,而是真實的生物學。
Part 1:背景知識——給剛入門的你
什麼是 TP53?
TP53(又稱 p53)被稱為「基因組的守護者」(Guardian of the Genome),是人體中最常被突變的腫瘤抑制基因,在所有人類癌症中,大約 50% 都有 TP53 突變。
TP53 蛋白的工作是:當細胞 DNA 受到損傷時,啟動修復程序,如果修不好,就啟動細胞凋亡(讓細胞自殺),防止損傷的細胞繼續分裂變成腫瘤。
關鍵結構特徵:TP53 是一個 四聚體蛋白——它需要四個 TP53 分子組裝在一起才能正常工作,這個特性直接決定了 DN 和 LOF 突變的差異。
TP53 蛋白有幾個重要的結構域(domain):
| 結構域 | 位置(大約) | 功能 |
|---|---|---|
| TAD(轉錄活化域) | N 端 | 啟動下游基因表達 |
| DBD(DNA 結合域) | 殘基 94–293 | 直接與 DNA 結合,辨識目標基因 |
| TET(四聚化域) | 殘基 319–360 | 讓四個 TP53 組裝在一起 |
| CTD(C 端調控域) | C 端 | 調控 DNA 結合活性 |
什麼是 Dominant-Negative(DN)vs. Loss-of-Function(LOF)?
這是理解整篇論文的核心概念,用一個比喻來說明:
想像一個划龍舟隊伍有四個人(= TP53 四聚體):
LOF 突變:其中一個人受傷退出,剩下三個人繼續划,船還是能動——只是少了一份力量,但不影響其他人的表現。
DN 突變:受傷的人堅持上船,不但自己不划,還故意把船槳往反方向划,導致整條船無法前進——一個人拖垮整支隊伍。
在分子層面:
- LOF:突變的 TP53 無法摺疊或無法結合 DNA,但不影響其他正常的 TP53 分子
- DN:突變的 TP53 仍然能參與四聚體組裝,但組裝後的四聚體無法正常結合 DNA,等於把正常的 TP53 一起拉下水
臨床意義:DN 突變跟 Li-Fraumeni 症候群(一種遺傳性癌症易感症候群)的更高外顯率有關,代表患者更年輕就會發病,且癌症種類更廣。
什麼是 AlphaMissense?
AlphaMissense 是 DeepMind(Google)在 2023 年發表的蛋白質致病性預測工具。它基於 AlphaFold 的架構,針對人類蛋白質組中所有可能的錯義突變(約 7,100 萬個),預測每一個突變是「良性」還是「致病」的。
重要限制: AlphaMissense 預測的是「致病性」(pathogenicity),但 不區分致病的機制。它會告訴你「這個突變可能致病」,但不會告訴你「它是透過 DN 還是 LOF 機制致病的」。
這正是本論文想解決的問題:能不能在 AlphaMissense 的基礎上,加入結構特徵來區分機制?
什麼是 FoldX?
FoldX 是一個計算蛋白質穩定性的經典工具。它可以:
- BuildModel:模擬一個突變,計算突變前後蛋白質穩定性的變化(ΔΔG,單位 kcal/mol)
- AnalyseComplex:計算蛋白質複合體中,某條鏈與其他鏈之間的交互作用能量
正的 ΔΔG 代表突變使蛋白質變得不穩定,負的代表更穩定。
本論文用 FoldX 計算了兩種 ΔΔG:
- 單體 ΔΔG(monomer):突變對單一 TP53 鏈的穩定性影響
- 界面 ΔΔG(interface):突變對 TP53 與其他鏈之間交互作用的影響——這是作者認為能區分 DN 和 LOF 的關鍵特徵
什麼是 Deep Mutational Scanning(DMS)?
跟上一篇文章一樣,DMS 是一次性測量蛋白質上幾乎所有可能突變效果的高通量技術。
本論文使用的是 Giacomelli et al. (2018) 的 TP53 飽和突變實驗數據。這個實驗在兩種細胞株(A549 和 H1299)中測量了 TP53 所有錯義突變的功能效果,根據兩種細胞株的不同表現模式,可以區分突變是 DN 還是 LOF。
經過嚴格篩選後,最終納入分析的是 105 個標記明確的突變:45 個 DN-only,60 個 LOF-only。
什麼是預先註冊(Pre-registration)?
這是本論文最特別的地方。
在傳統研究中,研究者可能會在看到結果後,調整分析方法或閾值,讓結果看起來更好——這叫做 p-hacking 或 HARKing(Hypothesizing After the Results are Known)。
預先註冊 是一種對抗這種偏差的方法:在跑任何模型之前,先把假說、分析方法和判定標準白紙黑字寫下來,甚至計算 SHA256 hash 鎖定文件內容,然後不管結果好壞都照實報告。
這篇論文設定了四個必須全部通過的「停止/前進」(stop/go)標準,結果三個失敗了——作者選擇誠實報告這個陰性結果,而不是換一個角度重新包裝成「正面」發現。這在學術界是很難得的。
Part 2:把生物問題轉成 ML 問題
定義標籤:什麼是 DN?什麼是 LOF?
不同於上一篇的 GPCR 研究需要自己設定連續值的閾值,這裡的標籤來自 Giacomelli et al. (2018) 的實驗設計——他們在兩種細胞株中測量了突變的效果:
- A549 細胞(有內源性 wild-type TP53):DN 突變能毒害內源性的正常 TP53,表現出明顯的生長優勢;LOF 突變因為不影響內源性 TP53 的功能,效果則小得多
- H1299 細胞(沒有內源性 TP53):沒有正常的 TP53 可以被毒害,DN 和 LOF 突變都表現為功能缺失
透過比較突變在這兩種細胞株中的差異表現——DN 突變在 A549 中特別突出,而在 H1299 中則不然——就可以區分 DN 和 LOF。
本論文採用 嚴格標記管線(strict labeling pipeline),只保留標記完全明確的突變:
TP53 所有錯義突變
→ 嚴格篩選(只保留 DN-only 或 LOF-only)
→ 105 個突變
→ 45 個 DN-only(42.9%)
→ 60 個 LOF-only(57.1%)
既有工具為什麼不夠用?
在動手建模型之前,作者先測試了兩個現成工具:
AlphaMissense:能預測致病性,但 不區分機制,它對所有 105 個突變都給出「致病」分數,但無法告訴你哪個是 DN、哪個是 LOF。
mLOF(Badonyi & Marsh):這是一個預測基因層級機制傾向的工具,但問題是——它對整個 TP53 基因只輸出 一個 結果(mLOF score = 0.567,「最可能是 LOF」),而不是對每個突變給出個別判斷。
所以,目前沒有開源的 per-variant 機制分類器可以用在 TP53 上,這就是本論文的出發點。
特徵設計:從結構中提取什麼資訊?
作者設計了兩組模型:
基線模型(Baseline,2 個特徵):
| 特徵 | 它在衡量什麼 | 來源 |
|---|---|---|
| am_pathogenicity | 這個突變有多「致病」 | AlphaMissense |
| monomer_ddG_2AC0 | 單體穩定性變化 | FoldX BuildModel |
完整模型(Full,6 個特徵):基線 + 4 個複合體特徵
| 特徵 | 它在衡量什麼 | 來源 |
|---|---|---|
| interface_ddG_A_vs_BCD | 鏈 A 與其他三條鏈的界面能量變化 | FoldX AnalyseComplex |
| interface_ddG_A_vs_DNA | 鏈 A 與 DNA 的界面能量變化 | FoldX AnalyseComplex |
| dist_to_DBD_contacts | 到 DBD 功能接觸位點的距離 | PDB 2AC0 結構計算 |
| dist_to_TET_assembly | 到四聚化域組裝殘基的距離 | 計劃中(但未成功實作) |
核心假說: 如果 DN 突變必須進入四聚體才能「搗亂」,那它們在蛋白質複合體界面上的能量特徵應該跟 LOF 突變系統性地不同——DN 突變應該保留界面結合能力(才能組裝),而 LOF 突變可能破壞界面。FoldX AnalyseComplex 正好可以量化這種差異。
結構覆蓋問題:43% 的突變沒有結構特徵
這裡有一個實務上很重要的問題:作者使用的結構 PDB 2AC0 只涵蓋 DBD 域(殘基 94–293),但 105 個突變中有 45 個(43%)落在這個範圍之外。
這些「範圍外」的突變,所有 FoldX 相關特徵都是 NaN,作者使用 中位數填補(median imputation)加上缺失指示變數(missingness indicator) 來處理:
from sklearn.impute import SimpleImputer
imputer = SimpleImputer(strategy="median", add_indicator=True)
X_imputed = imputer.fit_transform(X)
add_indicator=True 是一個有意為之的設計選擇——因為「是否為 NaN」本身就編碼了位置資訊(範圍外的突變 = NaN),作者刻意讓模型能看到這個模式,而不是隱藏它。這種透明性與稍後的 position-only 控制分析一脈相承。
預先註冊的 Stop/Go 標準
在跑任何模型之前,作者設定了四個標準,必須全部通過 才算「前進」(GO):
| 標準 | 閾值 | 含義 |
|---|---|---|
| C1:AUROC delta | > +0.03 | 完整模型必須比基線好至少 0.03 |
| C2:Bootstrap 95% CI | 下界 > 0 | 改善必須統計顯著 |
| C3:Feature importance | ≥ 1 個複合體特徵在前 5 | 複合體特徵必須真的有貢獻 |
| C4:Fold consistency | ≥ 7/10 次重複一致 | 改善必須穩定可重複 |
這是非常嚴格的標準設計——不是「任一通過」,而是「全部通過」。
Part 3:模型訓練與評估
兩種分類器的選擇
作者同時使用了兩種模型:
Logistic Regression(LR) 作為預先註冊的主要模型——簡單、可解釋、不容易過擬合。
Random Forest(RF) 作為非線性交叉驗證——能捕捉特徵間的交互作用。
from sklearn.linear_model import LogisticRegression
from sklearn.ensemble import RandomForestClassifier
from sklearn.model_selection import RepeatedStratifiedKFold
from sklearn.pipeline import Pipeline
from sklearn.impute import SimpleImputer
from sklearn.preprocessing import StandardScaler
# 主要模型:Logistic Regression
lr_pipeline = Pipeline([
('imputer', SimpleImputer(strategy='median', add_indicator=True)),
('scaler', StandardScaler()),
('clf', LogisticRegression(max_iter=2000))
])
# 交叉驗證模型:Random Forest
rf_pipeline = Pipeline([
('imputer', SimpleImputer(strategy='median', add_indicator=True)),
('clf', RandomForestClassifier(n_estimators=300, max_depth=4))
])
# 5-fold CV,重複 10 次
cv = RepeatedStratifiedKFold(n_splits=5, n_repeats=10, random_state=42)
結果:預先註冊的判定
| 模型配置 | 特徵數 | LR AUROC | RF AUROC |
|---|---|---|---|
| Position-only(控制) | 1 | 0.673 | 0.946 |
| Baseline(單體) | 2 | 0.892 | 0.924 |
| Full(預先註冊) | 6 | 0.904 | 0.956 |
| Full minus position(敏感性) | 4 | 0.901 | 0.941 |
預先註冊比較:Full vs. Baseline
Δ AUROC = +0.013
Bootstrap 95% CI = [−0.025, +0.055]
Fold consistency = 6/10
四個標準的判定結果:
| 標準 | 閾值 | 觀察值 | 通過? |
|---|---|---|---|
| C1:AUROC delta | > +0.03 | +0.013 | 未通過 |
| C2:Bootstrap CI 下界 | > 0 | −0.025 | 未通過 |
| C3:Feature importance | ≥ 1 | 腳本錯誤,無法評估 | — |
| C4:Fold consistency | ≥ 7/10 | 6/10 | 未通過 |
判定結果:STOP。 三個標準獨立失敗,複合體結構特徵沒有在預先設定的閾值上超越基線。
關於 C3 為什麼沒有評估:feature importance 的計算腳本因為 SimpleImputer 產生的指示變數數量與 feature_names 不匹配而報錯,作者選擇 不在事後修補腳本,理由是:判定結果已經由 C1、C2、C4 決定了,事後修補評估程式碼會打開「選擇性重新分析」的後門——這正是預先註冊要防止的事情。
這個決定在方法論上是正確的,即使它讓論文看起來「不完美」。
Part 4:兩個意外發現——為什麼「失敗」比「成功」更有資訊量
發現一:AlphaMissense 隱含了機制資訊(AUROC 0.892)
AlphaMissense + 單體 FoldX 這個只有兩個特徵的基線模型,在一個 它從未被訓練來做的任務 上達到了 AUROC 0.892。
為什麼這很令人驚訝?AlphaMissense 被訓練來預測的是「這個突變是否致病」,而不是「它透過什麼機制致病」。它不知道什麼是 DN 或 LOF。
那為什麼它能區分?作者提出了一個假說:
DN 突變可能承受比 LOF 突變更強的負向選擇壓力,原因是:一個 DN 等位基因可以毒害整個四聚體,所以它的破壞力遠大於只影響一個亞基的 LOF 等位基因。如果 AlphaMissense 透過序列上下文捕捉到了這種不對稱的選擇壓力,它的致病性分數就會 隱含地 與機制相關——即使機制標籤從未在訓練中使用過。
這是一個假說,不是結論,但實證觀察本身(AUROC 0.892)是確定的,而且據作者所知,這個數字從未在文獻中被報告過。
對未來研究的建議: 任何聲稱在 TP53 上做 DN vs. LOF 分類的新模型,都應該以 AM + 單體 FoldX(AUROC 0.892)作為最低基線來比較。
發現二:位置本身就是最強的預測器(AUROC 0.946)
這是更意外的發現。作者把「突變在蛋白質上的殘基位置」作為唯一特徵,用 Random Forest 來分類,結果 AUROC 達到 0.946——比用了 6 個精心設計的結構特徵的完整模型(LR AUROC 0.904)還高。
為什麼?看一下突變的分佈就明白了:
DN-only(45 個突變):
├── DBD 域(殘基 94–293):43 個(95.6%)
├── TET 域(殘基 319–360):0 個(0%)
└── 其他區域:2 個(4.4%)
LOF-only(60 個突變):
├── DBD 域(殘基 94–293):17 個(28.3%)
├── TET 域(殘基 319–360):37 個(61.7%)
└── 其他區域:6 個(10.0%)
幾乎所有 DN 突變都在 DBD,幾乎沒有 DN 突變在 TET;而 LOF 突變的大多數在 TET,一個 Random Forest 只要學會「如果位置在 TET 範圍 → LOF;如果位置在 DBD 範圍 → 可能是 DN」,就能拿到 0.946。
但這不是 bug——這是真實的生物學。
為什麼 DN 突變不會出現在 TET 域?用龍舟的比喻:要當「搗亂的隊友」,你首先得 能上船。TET 域負責的就是「讓四個人上船」的組裝過程,如果突變破壞了 TET 域,這個突變的 TP53 根本無法加入四聚體——它上不了船,自然就無法搗亂,只能被歸類為 LOF。
反過來,DBD 域負責的是「划船的方向」(DNA 結合),DBD 突變可以保留上船的能力(TET 域完好),但一旦上船就把方向帶偏——這就是典型的 DN 機制。
這個發現的啟示: 在 Giacomelli 嚴格標記的 TP53 資料集上,DN/LOF 的區分很大程度上是一個 位置分區 問題,任何新的分類模型都應該報告 position-only Random Forest 的效能作為 sanity check——如果你的模型沒有超越「只看位置」,那它可能只是在用更複雜的方式做同一件事。
為什麼 Logistic Regression 在 position-only 上只有 0.673?
Position-only 的 LR AUROC 只有 0.673,而 RF 有 0.946,差距巨大。原因是位置與標籤之間的關係是 非線性、非單調 的:
- 位置 94–293(DBD)→ 傾向 DN
- 位置 319–360(TET)→ 傾向 LOF
- 其他位置 → 混合
Logistic Regression 只能學到「位置越大/越小 → 越可能是某類」這種單調關係,它無法表達「中間一段是 DN,後面一段是 LOF」這種分段規則,Random Forest 可以,所以差距極大。
Part 5:為什麼 FoldX 界面能量沒有用?
這是一個值得深思的問題。作者的假說聽起來很合理:DN 突變保留界面結合能力,LOF 突變破壞界面——用 FoldX AnalyseComplex 來量化界面能量應該能區分兩者。
但結果是 沒有可偵測的額外貢獻,最可能的解釋是 資訊重疊:
FoldX 界面 ΔΔG 和 AlphaMissense 都在編碼「結構破壞」的某些面向,在只有 105 個樣本的資料集上,FoldX 能提供的額外資訊(超越 AM 已經捕捉到的)小到無法被偵測。
注意這不代表 FoldX 界面能量「沒有用」,它代表的是:在這個特定的任務上、對抗這個特定的基線、在 N = 105 的樣本量下,額外貢獻低於預設的偵測閾值。
統計功率分析也支持這個解讀:N = 105、類別比例 45:60 的情況下,每個測試 fold 只有約 21 個樣本,在 α = 0.05、power = 0.8 的條件下,能可靠偵測的最小 AUROC delta 大約是 +0.05 到 +0.07——比預設閾值 +0.03 還大,一個真實但微小的效果在這個設計下是看不見的。
附錄 A:Feature Engineering 實戰細節(進階)
以下內容適合有 Python 和結構生物學經驗、想深入了解或重現此研究的讀者,如果你只想了解研究的整體思路和結果,可以跳過。
FoldX 計算流程
FoldX 需要對 PDB 結構進行幾個步驟的處理:
# Step 1: 修復結構(補缺失側鏈、優化氫鍵等)
foldx --command=RepairPDB --pdb=2AC0.pdb1
# Step 2: 計算單體 ΔΔG(突變對單條鏈穩定性的影響)
foldx --command=BuildModel \
--pdb=2AC0_Repair.pdb \
--mutant-file=individual_list.txt
# Step 3: 計算界面 ΔΔG(鏈 A vs. 鏈 BCD)
foldx --command=AnalyseComplex \
--pdb=2AC0_Repair.pdb \
--analyseComplexChains=A,BCD
# Step 4: 計算界面 ΔΔG(鏈 A vs. DNA 鏈)
foldx --command=AnalyseComplex \
--pdb=2AC0_Repair.pdb \
--analyseComplexChains=A,EF
PDB 2AC0 是什麼? 這是 TP53 DBD(DNA 結合域)四聚體與 DNA 結合的晶體結構,由 Kitayner et al. (2006) 解析,包含四條蛋白質鏈和 DNA,涵蓋殘基 94–293。
AlphaMissense 分數提取
import pandas as pd
# 從官方 Zenodo 批量檔案中提取
am = pd.read_csv('AlphaMissense_aa_substitutions.tsv.gz',
sep='\t', comment='#')
# 篩選 TP53(UniProt P04637)
tp53_am = am[am['uniprot_id'] == 'P04637']
# 與突變列表合併
merged = variants.merge(tp53_am,
on=['position', 'alt_aa'],
how='left')
缺失值處理的細節
from sklearn.impute import SimpleImputer
# add_indicator=True 會為每個有 NaN 的特徵列
# 額外產生一個二元指示列(0/1)
imputer = SimpleImputer(strategy='median', add_indicator=True)
X_imputed = imputer.fit_transform(X)
# 例如:原本 6 個特徵 → 最多變成 6 + 6 = 12 個
# (實際上有 5 個 2AC0 衍生特徵有 NaN,
# 所以是 6 + 5 = 11 個)
為什麼不刪除 NaN 行? 因為 45 個落在 2AC0 範圍外的突變中有 43 個是 LOF——如果刪除它們,就會嚴重改變類別比例和分類難度。
Bootstrap 信賴區間計算
import numpy as np
from sklearn.metrics import roc_auc_score
def bootstrap_auroc_delta(y_true, y_pred_full, y_pred_base,
n_iterations=1000, random_state=42):
rng = np.random.RandomState(random_state)
deltas = []
for _ in range(n_iterations):
# 有放回地重新抽樣
idx = rng.choice(len(y_true), size=len(y_true), replace=True)
y_boot = y_true[idx]
# 跳過只有一個類別的 bootstrap 樣本
if len(np.unique(y_boot)) < 2:
continue
auroc_full = roc_auc_score(y_boot, y_pred_full[idx])
auroc_base = roc_auc_score(y_boot, y_pred_base[idx])
deltas.append(auroc_full - auroc_base)
deltas = np.array(deltas)
ci_lower = np.percentile(deltas, 2.5)
ci_upper = np.percentile(deltas, 97.5)
return np.mean(deltas), ci_lower, ci_upper
附錄 B:重現這個研究(進階)
環境設置
# 建議使用 conda 環境
conda create -n dnsense python=3.10
conda activate dnsense
# 安裝核心套件
pip install pandas numpy scikit-learn==1.4 openpyxl matplotlib seaborn
# FoldX 需要從官方網站下載(需要學術授權)
# https://foldxsuite.crg.eu/
資料取得
- TP53 DMS 資料: Giacomelli et al. (2018) 的 Supplementary Materials(DOI: 10.1038/s41588-018-0204-y)
- AlphaMissense 分數: Zenodo 批量檔案(DOI: 10.5281/zenodo.8208688)
- PDB 結構: 2AC0(DBD 四聚體 + DNA)、1OLG(TET 四聚體)從 RCSB PDB 下載
- mLOF 工具: https://github.com/badonyi/mechanism-prediction
延伸閱讀
如果你對這個領域有興趣,以下資源可能有幫助:
- AlphaMissense: Cheng et al. (2023) Science. 蛋白質致病性預測的代表性工作。
- mLOF 機制預測: Badonyi & Marsh (2025) Nature Communications. 基因層級的機制分類。
- TP53 飽和突變: Giacomelli et al. (2018) Nature Genetics. 本論文的資料來源。
- FoldX: Delgado et al. (2019) Bioinformatics. 蛋白質穩定性計算工具。
- DN vs. LOF 的結構基礎: Gerasimavicius et al. (2022) Nature Communications. 不同突變機制對蛋白質結構的影響。
- 預先註冊在計算生物學中的應用: 這是一個仍在發展中的領域,但臨床試驗和心理學已經廣泛採用。
關於作者
我是 Po-Chun Chen,獨立研究者,研究方向為計算生物學與資訊安全,這是我以獨立研究者身份發表的第二篇論文。
如果你有問題、想討論,歡迎透過 [email protected] 聯繫我。
論文連結:Chen, P.-C. (2026). Pre-registered evaluation of complex-aware structural features for TP53 dominant-negative versus loss-of-function classification: no detectable gain over an AlphaMissense plus monomer FoldX baseline. Zenodo. https://zenodo.org/records/19494246
Member discussion